In the News
Mr Henderson’s gene therapy work featured in national media
Why choose Mr Henderson for inherited retinal disease?
One of the UK’s leading retinal gene therapy specialists, Mr Henderson was instrumental in commissioning the NHS RPE65 Luxturna programme at Moorfields and GOSH — the first approved gene therapy for blindness in the UK. He serves as Chief Investigator for the Phase 4 Luxturna study and was Principal Investigator on the world’s first CLN2 ocular gene therapy trial. For families affected by inherited retinal disease, he offers the most advanced therapeutic options available in the UK. Book a consultation with a specialist in London.
Inherited Retinal Disease
Inherited retinal dystrophies are a group of genetic conditions causing progressive loss of retinal function. Accurate diagnosis, genetic counselling, and specialist surveillance are the cornerstones of management — and for some conditions, gene therapy is now a reality.
What Are Inherited Retinal Dystrophies?
Inherited retinal dystrophies (IRDs) are caused by mutations in any of over 300 genes that are essential for the development or function of the photoreceptors and retinal pigment epithelium. They are the leading cause of visual impairment in working-age adults in the UK.
The pattern of visual loss, age of onset, and rate of progression vary enormously depending on the underlying genetic cause. Some conditions primarily affect peripheral vision (rod dystrophies), others affect central vision (cone dystrophies), and many affect both.
“An accurate genetic diagnosis is the foundation of everything — it informs prognosis, guides surveillance, enables genetic counselling for the family, and determines eligibility for emerging gene therapy trials.”
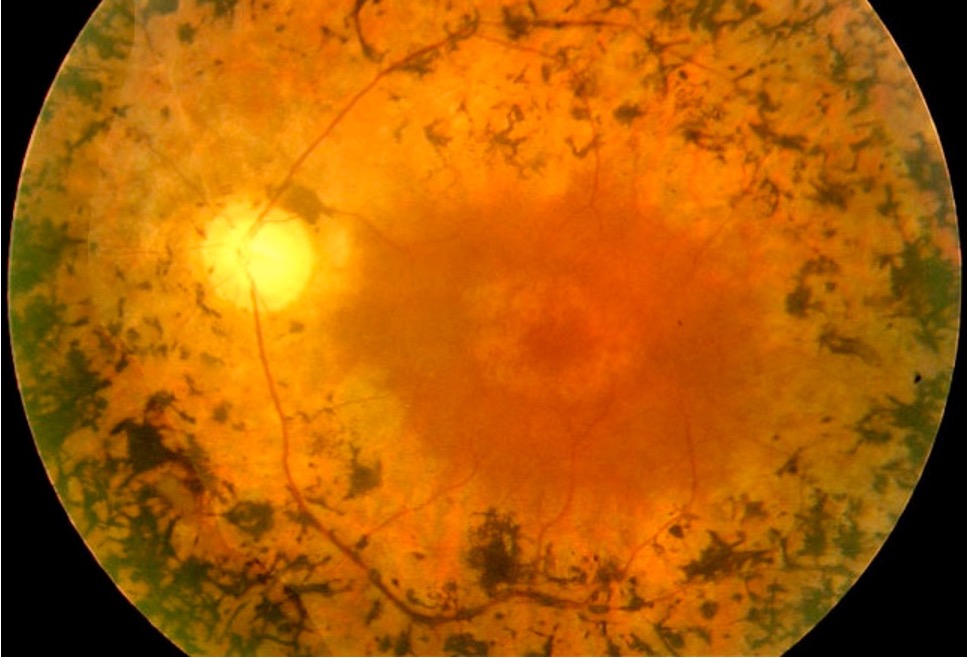
Common Inherited Retinal Dystrophies
Retinitis Pigmentosa
The most common IRD. Rod photoreceptors are primarily affected, causing progressive night blindness and loss of peripheral visual field, often with preservation of central vision until late stages.
Stargardt Disease
The most common juvenile macular dystrophy, caused by mutations in ABCA4. Affects central vision in the first or second decade of life, with relative sparing of peripheral vision.
Choroideraemia
An X-linked condition causing progressive degeneration of the choroid and retina. Affects males primarily, with night blindness and constricted visual fields progressing to severe visual loss.
Usher Syndrome
Combined retinitis pigmentosa and congenital sensorineural hearing loss. The most common cause of combined deaf-blindness. Several subtypes with different severity profiles.
Leber Congenital Amaurosis
A severe early-onset retinal dystrophy presenting at birth or in infancy. RPE65-related LCA is treatable with licensed gene therapy (Luxturna).
CRB1 Dystrophy
A severe form of early-onset retinal dystrophy with a distinct retinal appearance. Mr Henderson has a specialist research interest in CRB1-related disease at GOSH.
Paediatric & Adult IRD at GOSH and Moorfields
Mr Henderson holds a joint appointment at Great Ormond Street Hospital and Moorfields Eye Hospital, giving him unique expertise in inherited retinal disease across the full age spectrum — from early-onset conditions presenting in infancy to adult-onset dystrophies.
He is Clinical Lead for Ophthalmology at GOSH and holds an Honorary Associate Professorship at UCL-GOSH Institute of Child Health, where his research focuses on CRB1-related retinal dystrophies and gene therapy. He works closely with the Moorfields Inherited Eye Disease service to ensure patients have access to the most current diagnostic and therapeutic options.
Frequently Asked Questions
Your Journey from Diagnosis to Long-Term Care
Comprehensive electrophysiology, OCT, and genetic testing to confirm your specific condition and understand its trajectory — knowledge that is empowering, not frightening.
Mr Henderson leads the UK’s commissioned RPE65 gene therapy programme. Where gene therapy is appropriate, he will assess your eligibility and discuss what it can realistically achieve.
From gene therapy surgery to vision rehabilitation, Mr Henderson coordinates a multidisciplinary plan that maximises your functional vision for as long as possible.
Regular follow-up tracks disease progression and ensures you benefit from new treatments as they become available. Mr Henderson stays at the forefront of this rapidly evolving field.
“After years of uncertainty about my diagnosis, Mr Henderson gave me the clearest picture I’d ever had of what I had and what could be done. He treated me as an intelligent adult, explained the research honestly, and gave me back a sense of control. The gene therapy has genuinely changed my life.”
Gene Therapy in Focus
Current Developments in IRD Research
Gene therapy for inherited retinal disease has moved from laboratory promise to clinical reality. Below, Mr Henderson outlines where the science stands today — covering approved therapies, the current trial landscape, and what the next generation of treatments may look like for patients with conditions including retinitis pigmentosa, Leber congenital amaurosis, Stargardt disease, choroideraemia, and Bardet-Biedl syndrome.
Retina UK Professionals’ Conference 2025
Current developments in IRD research — an accessible overview of the gene therapy pipeline for allied health professionals, ECLOs and families.
Bardet-Biedl Syndrome Family Day
Mr Henderson speaks to families about the ocular features of BBS, what surveillance involves, and how gene therapy developments may affect those with this condition.
RNIB Connect Radio · May 2026
Listen: Saffie’s Story — Gene Therapy That Changed a Life
RNIB Connect Radio spoke with Saffie’s mother Lisa and Mr Henderson about Leber’s Congenital Amaurosis, what the Luxturna gene therapy procedure involves, and the difference it has made to Saffie’s vision and daily life. A 15-minute conversation that explains the treatment plainly and honestly.
Also available on Apple Podcasts
Where gene therapy stands in 2025
Voretigene neparvovec (Luxturna) remains the only licensed gene therapy for an inherited retinal dystrophy in the UK — approved for RPE65-related Leber congenital amaurosis and delivered at Moorfields and GOSH as the national commissioned programme. Mr Henderson serves as Chief Investigator for the Phase 4 study.
Beyond RPE65, an expanding pipeline of gene therapies is in active clinical trials for retinitis pigmentosa, Stargardt disease, choroideraemia, RPGR-related disease, and conditions including Bardet-Biedl syndrome and CLN2 Batten disease. Mr Henderson was Principal Investigator on the world’s first CLN2 ocular gene therapy trial.
For families with a confirmed genetic diagnosis, it is worth understanding whether your specific genotype has an active trial. An up-to-date assessment with a specialist will clarify your options — including access to trials not publicly advertised.
Arrange a Consultation
Mr Henderson personally leads all inherited retinal disease assessments and gene therapy surgery. His expertise in this field is unmatched in the UK.
To arrange an assessment for inherited retinal disease, please contact Alison Anscombe, Mr Henderson’s secretary:
+44 7974 015691 · alison.anscombe1@nhs.net
Or use the contact form on this website.